
Publication
International arbitration report
In this edition, we focused on the Shanghai International Economic and Trade Arbitration Commission’s (SHIAC) new arbitration rules, which take effect January 1, 2024.
Global | Publication | May 2016
As with the other process sectors, the life science industries (comprising the pharmaceuticals, biotech and medical technology industries) rely on the construction and operation of high value and complex process plant infrastructure.
One of the distinguishing features of the life sciences industries, in contrast to the wider process sector, is the robust, complex and rapidly evolving regulatory landscape to which they are subject. This undoubtedly impacts on the way in which process plant infrastructure will be procured and delivered in these vital sectors.
What merits particular attention from a regulatory perspective is the concept of Good Manufacturing Practices (GMPs). As part of the wider quality management system, GMPs set down regulatory guidelines on good practice for the manufacture of active pharmaceutical ingredients. GMPs represent the minimum standards that must be achieved before developer is granted a licence to manufacture.
Directly connected with this, and in addition to more general requirements around product safety, are requirements around anti-contamination procedures and controls and accordingly the reliance that these industries place on so-called ‘clean room’ technologies.
The interface between these clean room technologies and the process infrastructure they house creates particular issues to be considered when formulating contractual structures for the delivery of relevant process plant infrastructure.
In this article we will consider the key issues from a regulatory, risk allocation and contract structuring perspective that will be relevant for developers of process plant infrastructure when seeking to secure project realisation in accordance with their key time, cost and specification requirements.
Demographic changes in both developed and emerging markets are culminating in an increasingly ageing population globally. It is believed that this, along with growing populations and growing affluence in emerging markets, is likely to translate into increasing life sciences spend in the coming years.
“The EIU estimate increases in global pharmaceuticals sales by 6.9% per annum between 2014-18.1”
The development of new manufacturing and R&D facilities by life sciences companies directly or through partners is likely to increase as a result of a variety of factors:
While the industry outlook is generally encouraging, there are a number of key challenges facing those operating in the sector, such as (i) impending patent expiry cliffs for ‘blockbuster’ drugs, (ii) increased competition from generics, bio-similars and the emerging markets generally and (iii) governments and the other payers increasingly looking to control costs (e.g. through price controls).
So, despite the positives outlined above, it is likely that there will be those operating in the sector who may experience a squeeze on returns and profitability in the near to medium term.
It will therefore be essential for those developing new R&D and manufacturing capacity to carefully consider the types of issues raised in this article in order to properly manage the risks associated with infrastructure delivery:
In broad terms, GMP can be described as the minimum regulatory standards that a manufacturer must meet in its production and manufacturing processes in order to obtain and maintain a license to manufacture.
It will be helpful, for the purposes of this briefing, to provide a high level overview of the regulatory framework under which these requirements are implemented. More detailed and jurisdiction specific advice should of course be sought on a case-by-case basis.
At a global level, the World Health Organisation (WHO) has established its own GMP guidelines which tend to be applied mainly in developing countries. The WHO recommends several types of inspection for manufacturing facilities to establish and monitor GMP compliance and will make recommendations for regulatory actions in the case of noncompliance.
The WHO GMP guidelines and requirements
are often adjusted to meet local conditions and defer to the licencing, inspection and enforcement of the national regulatory body.
The GMP standards of the WHO can be found generally in the GMP standards of developed countries, where they typically form a sub-set of more detailed quality and safety assurance systems.
Under EU law, all manufacturers and importers of medicines located in the European Economic Area must hold a manufacturing licence.
Whilst the European Medicines Agency (EMA) oversees and coordinates compliance monitoring, the licencing regime is managed by the regulatory authorities of the individual Member States, which are responsible for issuing licences for activities taking place within their territories. In particular, the regulatory authorities are required to perform all necessary inspections to ensure that applicants for and holders of a manufacturing licence adhere to EU-level principles and guidelines of GMP.2
In the UK, the Medicines and Healthcare Products Regulatory Agency (MHRA) is the competent or relevant regulatory authority. In compliance with EU law, in order to make, assemble or import medicines in the UK it is necessary to first obtain a manufacturer’s licence.
The MHRA will only issue a licence to manufacture when it is satisfied, following an inspection of the site of manufacture, that the facility and the site conforms with GMP. Following issue of a licence, the MHRA will then perform periodic inspections to assess compliance with the relevant regulatory requirements, including GMP and compliance with the provisions of the manufacturing licence. Critical deficiencies identified during any such inspection may lead to revocation of the manufacturing licence and/or criminal sanctions.
In the US, whilst the activities of the Food and Drug Administration (FDA) are much broader than the activities of EMEA and the MHRA (i.e. not being limited to pharmaceuticals regulation alone), its role with regard to pharmaceutical regulation is broadly similar.
The FDA uses the term ‘current’ GMP (cGMP) in the context of the manufacture of pharmaceutical products to emphasise that manufacturers have to employ up-to-date technologies and systems to comply with regulation and to be licenced to market and have the right to manufacture.
The approval and licencing process for new drug and generic drug marketing applications includes a review of the manufacturer’s compliance with the cGMP. FDA inspectors will determine whether the developer has the necessary facilities, equipment and skills to manufacture the new product for which it has applied for approval. Decisions regarding compliance with cGMP regulations are based upon inspection of the facilities, sample analyses, and the compliance history of the developer.
The FDA can issue a warning letter or initiate other regulatory actions against a company that fails to comply with cGMP. Failure to comply can also lead to a decision by the FDA not to approve an application to market a drug and the withdrawal of the right to manufacture.
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) has representatives from the regulatory agencies and the pharmaceutical industries of the EU, the US and Japan. The ICH’s primary objective is the harmonisation of regulatory requirements related to quality, safety and efficacy of medical products and to support mutual recognition among the three regulatory authorities.
The key achievement of ICH has been the harmonisation of GMP among the relevant regulatory authorities through the Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. This was developed and recommended for adoption in the EU, Japan and the US in 2000.
Note that there is an important difference in perspective between the EU/Japan approach and the US, with regard to demonstrating compliance. For the EU/Japan, more reliance is placed on written procedures (such as SOPs), as sufficient evidence of GMP compliance. For the US, such procedures are only a beginning of such evidence with batch records, in-process sampling, and final release data as requisite endpoints of showing GMPs being met.
We will later go on to consider how the risk of GMP compliance in this regard may be managed through the supply chain with particular focus on the testing and handover of the plant.
It is worth noting that in Europe, recent updates to guidance in respect of qualification and validation requirements3 have been introduced to update and modernise the relevant regime and to more generally bring the guidance in line with FDA Guidance on Process Validation.
Qualification requirements typically apply to equipment, facilities, utilities or systems and fall within the following key categories:
Installation Qualification (IQ) – this will include verification that the relevant item conforms with the specification, that there has been correct installation of components, instrumentation, equipment, pipe work and services against the engineering drawings and specifications and that there has been verification of the materials used in construction.
Operational Qualification (OQ) – this will include verification that the relevant item achieves the specified operational requirements (throughput, power consumption, confirmation of upper and lower operating limits).
Performance Qualification (PQ) – this will include the testing of the item as per the OQ, but this time at full load/ throughput. So, the tests will use manufacturing materials in a ‘live’ operational environment.
The categories above and the criteria under each category will vary depending on the relevant project circumstances. The qualification requirements will typically be performed in the sequence above but there may be justification, for instance, in running the PQ at the same time as the OQ and process validation.
The qualification tests may also be supplemented with design qualification tests, factory acceptance testing and site acceptance testing.
Process validation is documented evidence that the process, when operated within established parameters, can perform effectively and reproducibly to produce a medicinal product meeting its predetermined specifications and quality attributes.4
The traditional validation process involves a number of batches of the finished product being manufactured under routine conditions to confirm reproducibility. In contrast, continuous process verification provides for the manufacturing process performance to be continuously monitored and evaluated. The continuous validation approach may be used where it has been established during development that the relevant control strategy provides a high degree of assurance of product quality. Nevertheless, the continuous validation process may be used in addition to or instead of the traditional validation process, this is often referred to as the hybrid approach to process validation.
Ongoing process validation through lifecycle is a requirement under the FDA and now the EMA guidelines. This is in addition to the traditional, continuous or hybrid approaches that may be employed. The rationale here is perhaps obvious, manufacturers should monitor product quality to ensure that a state of control exists throughout the product lifecycle. The requirements in this regard will be dictated on a case-by-case basis and may change through the product lifecycle as the level of process understanding and/or plant performance may change. This is clearly an operational issue although we consider below if and how this may remain a construction risk given that any failure may arise from defects in the plant.
A failure to achieve and demonstrate compliance with the relevant GMPs will give rise to liabilities beyond those merely connected to construction cost overrun and delay. A refusal to licence or enforcement by regulators against an existing licence can have a catastrophic effect on developers, impacting on their revenue and profit lines and possibly leading to adverse media coverage, brand damage, market share loss, and considerable liability costs.
Compliance with the relevant GMP standards must therefore be carefully managed through the procurement of new R&D or manufacturing capacity. These obligations of course need to be managed through the full life of the plant meaning that continued regulatory compliance during operations remains an absolute requirement that must be addressed contractually or otherwise by any developer.
Of course, it must be remembered that GMPs represent minimum standards only. Most life sciences companies will seek to adhere to GMPs but will also look to overreach these requirements with their own best practice standards. The principles above around considerations in procurement strategy should apply equally to these enhanced standards. We consider further below how GMP compliance may be appropriately managed through the construction supply chain.
Also comprised within GMP are requirements with regard to anti-contamination procedures and controls and accordingly the reliance that these industries place on so called ‘clean room’ technologies.
Use of this technology gives rise to certain interface issues that requires attention from a contract structuring perspective. These are discussed next along with how GMP compliance may be appropriately managed through the construction supply chain.
When we refer to process technology, we are referring to the technology used to facilitate:
Primary and secondary processing may take place on the same site, adjacent sites or on completely different sites.
In contrast to the process technology, the cleanroom technology is the environment in which the process technology is housed.
A clean room is an area of the facility in which the concentration of airborne particles is controlled to specific limits. The level of control will be dependent on the particular standards required.
In order to control contamination there must be control of the entire environment. Air flow rates and direction, pressurisation, temperature and humidity all need to be properly and effectively controlled and often central to the process is the use of specialised filtration systems such as High Efficiency Particulate Air Filters.
These specifications and developer requirements must be clearly defined at the outset of any project to avoid quality failure, developer disappointment and disputes between the developer and its supply chain.
The increase in regulatory interest in life sciences globally mentioned above, is fuelling the rapid growth in the cleanroom technology market globally. Projections suggest a growth rate in the market of 5.2% per annum, taking the value of this market to approximately US$4.29bn by 2020. This growth is also being further stimulated through increased demand for sterilised pharmaceutical formulation and the development of new biologics.5
A key issue for stakeholders in the life sciences industries will be what is perceived to be the interface risk arising between:
When we talk of interface risk we are referring to the risk, for example, of:
Where such risks cannot be or are not allocated to and absorbed by the supply chain, they will tend to rest with the developer. Accordingly, this will open the developer up to time and cost claims from the supply chain should the relevant interface risk materialise.
It is perhaps surprising that few contractors operating in the sector offer full design and construction capabilities for both process and clean room technologies such that they are prepared to assume full risk in delivery of all aspects of the project including absorbing the interface-type risks identified above. Given the level of interface involved between the individual facets of a typical project in the life sciences sector, this would surely be a big selling point for any contractor and would be likely to enable it to offer a price advantage over its competitors.6 Instead, a contractor offering a turnkey solution without full in-house capability is likely to be a more expensive proposition given that it will tend to price the risk it assumes in third party subcontracts for components outside of its own competencies.
This feature has tended to move the life sciences industries more towards a multi-contracting approach (that we describe below), since any single contractor may be unable or reluctant to assume full integration risk between the different process technologies particularly involving novel elements and between the process technologies and the cleanroom technology.
This is of course not to say that the single contractor EPC solution (also described below) has not been successfully deployed in these sectors. Developers should be aware however that the assumption by the contractor of a high level of interface risk is likely to have a material impact on the overall outturn cost.
Following completion of conceptual design, developer focus should be on the selection of an appropriate contract delivery structure. These considerations should take place before the commencement of detailed design. Experience shows that detailed design will tend to influence how contract delivery will be structured which will of course narrow developer choice and the opportunity for flexibility in procurement.
As a practice, we expend a significant portion of our time with our clients during this period of project formulation. It is important that the developer achieves its required position with regard to risk allocation and pricing and all potential options should be reviewed and analysed during this predetailed design phase on a cost-benefit-analysis basis.
Experience shows that a lack of detailed considerations at these early stages by the developer and its advisers/ consultants defining the specification and subsequently structuring the contract delivery structure can often lead to problems later. These problems can often strain relations between the developer and the supply chain leading to dispute. Ultimately, the risk to developers arising from a lack of planning is the potential for time and cost overrun risk exposure.
We provide below a high level description of some of the key contract options and issues to be addressed in light of matters already considered in this briefing.
The multi-contracting model will see the developer identifying the individual works and supply packages, procuring the contractors and suppliers, entering into contracts with them and managing itself the interface (design, programme, integration etc.) between the different packages and the achievement of the overall global project targets – such as planned budget, planned completion and planned date of full operations (the Key Project Targets).
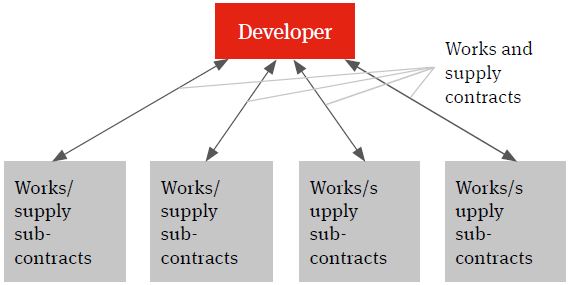
Figure 1. Multi-contracting solution
There are number of reasons why the developer may choose a multi-contracting solution.
Given the high degree of interface risk involved between the different facets of a typical project in the life sciences sectors, there may be a more limited number of contractors able and willing to assume full risk in delivery of the project on time, on budget and to a required technical and performance specification (often referred to as a ‘wrap’ of construction delivery risk). Contractors prepared to offer this wrap of construction delivery risk may in return include significant risk contingencies in their price for the works. This may impact on the developer’s returns and even the economic viability of the project. In contrast, the multi-contracting option is often seen to offer price advantages.
A multi-contracting approach tends to provide the developer with a degree of flexibility both in design development and in the procurement of the works and supply packages. Where a single contractor EPC solution is selected (see below), the contractor will typically agree to fix its price for project delivery based on known developer requirements at contract signature and there will be little scope for the developer further influencing design direction or procurement choice going forward (at least not without it having a significant price impact for the developer).
If a multi-contracting solution is selected, experience shows that the developer will require an experienced and wellresourced internal team to manage the procurement and the delivery of the works.
It may be the case that such resource and experience is not available to the developer and there may be the need to employ an experienced project management consultancy to act as the ‘developer team’. Obviously, this team should not be left to second guess developer requirements and so there must be clear lines of communication, such that developer requirements and specifications are clearly communicated and understood at project inception and throughout implementation.
The multi-contracting route can be contrasted with a single contractor turnkey solution under which the relevant contractor will wrap construction delivery risk for a fixed price. This model is often referred to as an engineer, procure and construct (EPC) model.
Whilst the developer will procure the relevant contractor and contract directly with it, the wrap of construction delivery risk being provided by the EPC contractor will (subject to the developer knowing specifications which can be priced by the contractor) typically see it assuming responsibility for identifying the individual supply and works packages, procuring the contractors and suppliers, entering into the individual works and supply contracts, managing the interface risk highlighted above (both practically and from a risk and a liability perspective) and for the achievement of the Key Project targets.
Often, the name on the front cover of a contract doesn’t reflect the content and minor deviations from key requirements can mean that unintended time and cost risk may filter back to the developer. Diligence should be exercised to ensure that the contract delivers the expected risk allocation, so as to avoid nasty surprises for the developer during project implementation.
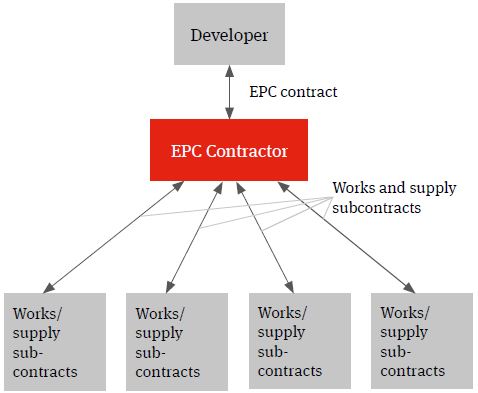
Figure 2. EPC solution
There are of course a number of hybrid contractual structure models which may also be considered by the developer.
Whilst the multi-contracting solution may give rise to capital cost savings when compared with a single contractor EPC solution, it is likely that greater amounts of risk are retained and need to be managed by the developer.
It is the means by which these risks are managed by the developer that is critical in achieving project success.
The availability of an experienced and well-resourced internal developer team or an experienced project management consultancy will go some way to assisting the developer in managing the retained risks referred to above and which are connected with a multi-contracting solution.
Experience shows however that the ability to manage retained risks can be enhanced significantly through the use of an engineer, procure and construction management (EPCM) contractor. We have seen the benefits that use of an EPCM solution can have in the realisation of process plant infrastructure delivery across a number of sectors for developers procuring infrastructure delivery on a multicontracting basis.
An EPCM contract is essentially a professional services appointment under which the EPCM contractor’s services will usually be limited to the production of detailed design and the procurement, construction management and coordination of the works and services necessary to deliver the project.
It is important to recognise that it is not a contract for the carrying out of construction works.
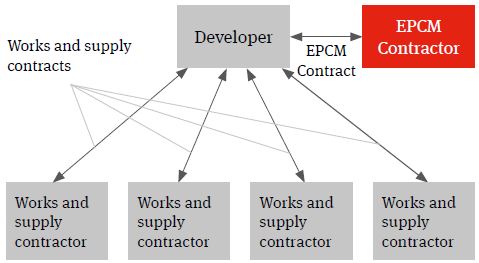
Figure 3. EPCM solution
The EPCM Contractor will contract directly with the developer and it is usually the developer that contracts with the works and supply package contracts (procured and managed by the EPCM contractor) not the EPCM contractor. The authority of the EPCM contractor to act on behalf of the developer and to manage the works and services will usually be documented in the EPCM contract and acknowledged in the works and services contracts.
Whilst often confused with the EPC contracting solution (mainly due to use of a very similar acronym), the EPCM and EPC contracting solutions are very different in terms of the nature of the obligations undertaken and the risk allocation assumed by the respective contractors. In contrast to the EPC model, which is based on risk transfer and more limited client/contractor interaction, the EPCM ethos is more about collaboration and cooperation and less about risk transfer.
The EPCM contractor will not backstop project delivery risk and, importantly, the achievement of the Key Project Targets. The EPCM contractor will however be responsible for managing the same on behalf of the developer but the levels of liability assumed by the EPCM contractor for failing in the performance of its obligations will typically be more limited (e.g. 10-20% of the EPCM services fee). If there is significant failure in the achievement of the Key Project Targets, the ‘buck’ will ultimately stop with the developer.
Whilst reputation in securing project delivery will be an important factor for the EPCM contractor, the developer will need to consider and implement contractual mechanics to appropriately incentivise the EPCM contractor to effectively and proactively manage and control project delivery and the achievement of the Key Project Targets.
The approach typically adopted under an EPCM solution relies on an incentive structure which provides positive (and sometimes negative) incentives to the achievement of the Key Project Targets and other associated targets e.g. health and safety performance, meeting the EPCM budgeted price etc. These provisions are generally bespoke and may be underpinned by fairly complex calculations but equally can be fairly straightforward depending on the approach preferred by developers.
Since the EPCM contractor will typically set the Key Project Targets and other associated targets through its procurement and programme planning obligations, appropriate due diligence will need to be undertaken by the developer team to establish that these are sensible in the context of the project and the relevant incentive payments/penalties proposed.
While assistance and support from a highly experienced EPCM contractor will be very important, it will not provide an ultimate backstop for a failure in achieving the Key Project Targets. Since these risks may then ultimately rest in the most part with the developer, it will be necessary for the developer to consider and plan for how these risks will be mitigated and/or managed. The developer should therefore also look to consider the following as part of risk planning:
The terms on which the works and services contracts are procured under EPCM or other multi-contracting model will remain very important. Where possible, these should be procured on a fixed price basis and should be on robust terms backed by a comprehensive security package arrangement. Whilst procurement will be led by the EPCM contractor, the developer will want to keep a very close eye on this and the forms of contract used and it would be sensible to include template forms in the EPCM contract terms.
The process plant sector as a whole tends to use an array of different standard form contracts. The most popular, in our experience, include the forms produced by the Institute of Chemical Engineers (IChemE), the forms produced by Fédération Internationale des Ingénieurs-Conseils (the International Federation of Consulting Engineers) (FIDIC) and the forms produced by the American Institute of Architects (AIA). They tend to be known by contractors operating in the sector and tend to be an acceptable starting point, with different ‘books’ being produced for different types of work and pricing structures. It is always sensible to harmonise the forms used across the project such that the different contracts work well together and this will of course also enable more efficient management of the individual packages.
Developers should look to harmonise dispute resolution procedures across the contracts and in particular, to provide for the right consolidate claims to enable it to bring related disputes under the same umbrella to avoid the inefficiency of multiple claims running at the same time on the same or connected issues. This may also be achieved through the interface arrangements described below.
The developer, along with their advisers or the EPCM contractor, may also propose bespoke forms of contract which may be another acceptable starting point. Bespoke forms may however on occasion be viewed with mistrust by contractors and suppliers and may be seen as a venture into the unknown when compared with the industry standard forms identified above. Unless properly managed, this approach could delay the procurement timetable.
An understanding of the risk allocation under the form of contract proposed will be important for the developer and will shape the need (as applicable) for amendments to the contract form proposed.
Developers may look to establish, where possible, an ‘EPC island’ approach under the EPCM structure. The idea here is that identified and discrete sections of the project will be delivered and ‘wrapped’ by a single contractor and this will assist in reducing the level of interface risk being retained by the developer. This is often seen for key aspects of the process technology or ancillary facilities such as waste water treatment or power.
If an EPC island approach is to be adopted, care should be taken to limit the level of design development in relation to the relevant package(s) during the pre-procurement phase. In our experience, EPC contractors may be reluctant to assume risk in detailed design produced by third parties (i.e. the EPCM contractor), at least not without pricing on a contingent basis the risk of accepting such design. There is of course a balance to be struck between the developer having clarity on the specification required and the exposure it can accept to the cost premium likely to be charged for the EPC contractor in assuming risk in third party design.
We have mentioned interface management contracts above in the context of interface risk. The use of these arrangements will represent a fairly innovative approach under which the works and supply contractors on the project (or for a discrete aspect of the project having high degrees of interface) will enter into a multi-party interface agreement.
A ‘collaborative’ or ‘alliancing’ approach has widespread use across the construction industry and is of great value in that it encourages and incentivises cooperation and coordination between individual contractors in the supply chain. The approach advocated here takes the alliancing approach one step further, hardwiring some of the key principles contractually between supply chain members.
An interface agreement should seek to establish how the parties will cooperate with one another, how they will resolve disputes and importantly it will seek to allocate the risk (time and cost) of managing interfaces to the supply chain.
We have experience of successful implementation of these types of bespoke arrangements on high value multicontracting process plant projects and would suggest that they represent a ‘high water mark’ in terms best practice in managing infrastructure delivery under an EPCM contract solution.
The satisfaction of relevant regulatory requirements and the securing of a licence to manufacture should ideally be a condition of hand over of the plant or a section (as the case may be). It will not usually be appropriate for the developer to define these requirements in the specification, as this may create a risk of developer error or the risk of interpretational arguments undermining the position that the developer thought it was achieving.
Works delivery on a multi-contracting basis may present challenges in this regard (i.e. no one contractor able on its own to satisfy the relevant requirements for licensing). The solution is the implementation of a fully integrated testing regime across all relevant works packages. The obvious place for this to be managed and for risk to be allocated is under the terms of multi-party interface agreement of the type discussed above.
It is important to recognise that in making regulatory compliance for full commercial operation a condition of hand-over, the risk of delay to commercial operations is transferred to the contractors, typically through payment of delay damages. These damages will represent important leverage over the contractor(s) and will incentivise the contractor(s) to do all that is necessary to remediate the relevant failures promptly in order to mitigate the relevant exposure.
Whilst the developers may wish to retain a right to reject the plant in these circumstances, this is unlikely to be an appropriate remedy in most circumstances as it must be weighed against the losses to the developer arising from a delay or failure in getting a new product to market.
Contractors will usually look to limit their delay damages exposure to an agreed cap. It will be important that the cap on delay damages is appropriately sized in view of the developer’s costs and losses arising out of the delay up to an agreed long stop date. It should be supplemented with a right to terminate the contract when the cap is reached. Such a termination right in these circumstances (whilst unlikely to be used) will provide important further leverage to secure an extension to the delay damages liability sub-cap.
The approach suggested above will involve certain operational tests being included in the pre-hand over requirements (e.g. Operational Qualification and Performance Qualification). This approach may require interface between the contractors and the operational contractors and this will need to be carefully managed contractually to ensure that acts or omissions of the operational contractors cannot derogate away from the responsibility of the contractor(s) to fully satisfy the hand over requirements.
Whilst indefinite retention of security would not be appropriate in respect of the on-going process validation as described above, it may be appropriate however for security in this regard to be held for a fixed period post-handover of the works until the first round of regulatory inspections have been completed (e.g. in relation to on-going process validation). The release of security should not however release the relevant contractor from liability to the extent that it otherwise continues under the terms of the relevant contract.
Outside of the absolute requirements arising from regulation, the developer will also want to establish performance of the plant against key performance indicators post-handover.
Typically, the contracts for plant delivery will provide for certain performance guarantees in respect of the key performance indicators (power consumption, throughput, availability) which are tested over a prolonged period of full operations (six months to two years). Any failure to demonstrate the guaranteed levels of performance will give rise to an obligation on the part of the relevant contractor to pay a performance liquidated damages to compensate the developer for its costs/losses arising from the performance shortfall for an agreed period. Since contractors again may seek to limit their performance damages exposure to a capped amount, in order to ensure that the performance damages provide for an adequate remedy, the developer should either look to:
Experience shows that this is the most important period in the construction phase of a project.
If a multi-contracting solution is to be adopted, planning the management of retained developer risks will be of paramount importance. We have identified in this article a number of key methods that may be adopted to manage and mitigate the relevant risks. There is however no ‘one size fits all’ magic solution and each project must be considered on its particular facts such that the optimum project delivery solution may be adopted achieving the developer requirements.
A lack of attention in this regard can often mean the difference between delivery of process plant infrastructure on time, on budget and to a required technical specification in compliance with regulatory requirements and an expensive project failure.
Publication
In this edition, we focused on the Shanghai International Economic and Trade Arbitration Commission’s (SHIAC) new arbitration rules, which take effect January 1, 2024.

Publication
On September 18, 2024, the "Decree amending the list that sets forth goods whose import and export are subject to regulation by the Ministry of Energy" (the "Decree") was published in the Federal Official Gazette.
Subscribe and stay up to date with the latest legal news, information and events . . .
© Norton Rose Fulbright LLP 2025
